
化学者にとって分子の励起エネルギーは分子の色彩や吸収スペクトルを知るうえで非常に重要です。励起エネルギーを知る際に最も利用される計算手法がTD-DFT(時間依存のDFT法)です。
「励起エネルギーを計算するのに、なぜ時間依存が必要?」と思う人も多いのではないでしょうか?その答えは、光励起の際に、分子は周期的に振動する電場を感じるからです。
このページでは、TD-DFTで分子の励起エネルギーが計算できる理由とそこに必要な3つの手続きを解説し、最終的に詳細なTD-DFTの表式までご紹介します。
- ホルムアルデヒドの励起エネルギーの計算例
- TD-DFTで励起エネルギーを知ることができる理由
- 励起エネルギーを知るための3つの手続き
- 水の励起エネルギーを固有値方程式を解いて求める

【具体例】ホルムアルデヒドの励起エネルギー
初めに、ホルムアルデヒドの例を見てみましょう。ホルムアルデヒドは最も低い励起エネルギーとして$4.1~4.2\text{eV}$(実験値)を持っています(H. Nakatsuji et al, J. Chem. Phy., 1989, 75, 2952)。
ホルムアルデヒドの励起エネルギーをTD-DFT法の量子化学計算で再現してみましょう。TD-DFTは無料ソフトウェアのGAMESSを利用すると簡単に実施でき、次のように励起エネルギーを求めることができます。
-------------------
SINGLET EXCITATIONS
-------------------
STATE # 1 ENERGY = 4.175510 EV
OSCILLATOR STRENGTH = 0.000000
LAMBDA DIAGNOSTIC = 0.513 (RYDBERG/CHARGE TRANSFER CHARACTER)
SYMMETRY OF STATE = A2
EXCITATION DE-EXCITATION
OCC VIR AMPLITUDE AMPLITUDE
I A X(I->A) Y(A->I)
--- --- -------- --------
5 9 -0.045685 0.001368
8 9 0.999028 -0.037909
8 13 0.030988 -0.000890TD-DFTによると、第一励起のエネルギーとして$4.175510\text{eV}$の結果となっており、実験値$4.1~4.2\text{eV}$に則した数値を与えることに成功しています。
さらに、「EXCITATION AMPLITUDE」の場所を見ると、値$0.99$を持っているHOMO軌道$\phi_8$からLUMO軌道$\phi_9$へ電子遷移がメインで寄与しつつ、$\phi_5\to\phi_9$や$\phi_8\to\phi_{13}$への電子励起も寄与することが示されています。
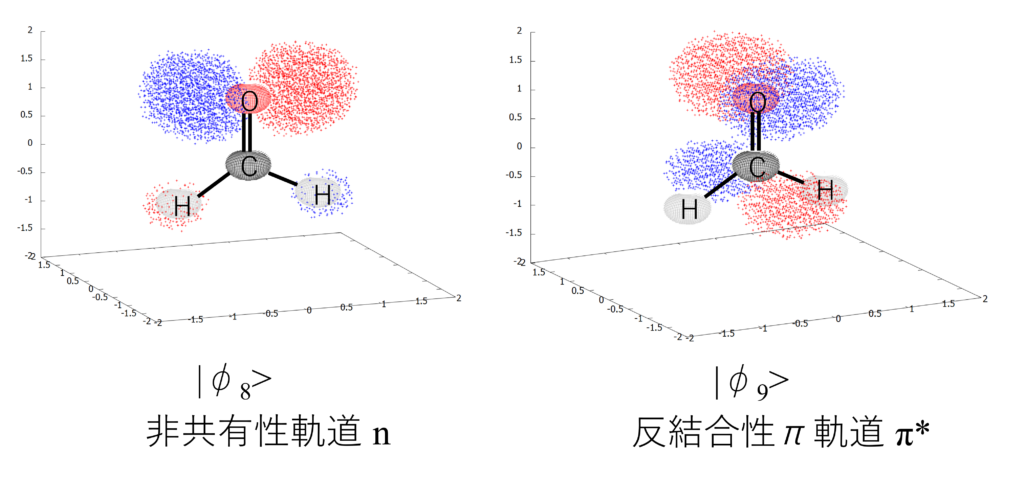

「DE-EXCITATION AMPLITUDE(脱励起強度)」は何を表しているの?

脱励起は励起による密度変化へ影響を与える高次の効果だよ。値も小さいから無視する場合(Dancoff近似)もあるよ
なぜTD-DFTで励起状態が得られるのか
TD-DFT(Time-Dependent DFT)とは「時間依存の」密度汎関数法です。では、なぜ密度汎関数法を時間依存させると分子の励起状態が得られるのでしょうか?
理由は、「分子に対して光を当てた際に分子の電子状態がどのように変化するか」で励起状態を調べているからです。

光は電磁波だから短時間で周辺の電界が激しく振動しているよね
可視光(数$\text{eV}$程度)でも$1$秒に$10^{15}$回($\omega=10^{15}\text{Hz}$)以上で振動しているよ
分極$p$を持つ分子に光を当てると、振動電場${E}\cos(\omega{t})$によって分子にエネルギー$\delta{v(t)}=pE\cos(\omega{t})$が与えられ、電子密度は基底状態$P^{(0)}$から$P^\prime(t)=P^{(0)}+\delta{P}\cos(\omega{t})$へ変化すると考えます。この$P^\prime(t)$を励起状態の電子密度と捉えることで、時間依存の密度汎関数法「TD-DFT」で励起状態の電子状態を計算するのです。
ということは、DFTにこだわらなくてもHFでも励起エネルギーを計算できるということだね。
ページの最後ではTD-DFTではなく、TD-HFで水の励起エネルギーを計算しているので、ぜひ見てみてください。
TD-DFTで励起エネルギーを知る手続き
次に、TD-DFTではどのようにして励起状態を得るのでしょうか。主に3つの手続きを踏んで励起状態と励起エネルギーを計算します。
- 光エネルギーを「摂動」と捉える
- 基底状態の計算で得られた分子軌道をもとに励起状態を複数の電子配置で表現する
- 励起状態のエネルギーは発散しないとする
【ステップ1】光エネルギーと電子密度が比例で応答すると考える
まずは、光によるエネルギー$\delta{v}=f\cos(\omega{t})$が分子に与えられた際に、比例して電子密度の変化$\delta{P}$が得られると考えます。
つまり、基底状態の電子密度を$P^{(0)}$、励起状態の電子密度を$P^\prime=P^{(0)}+\delta{P}$としたとき、$\delta{P}=\alpha\times\delta{v}$の関係があると仮定します。
エネルギー$\delta{v}=f\cos(\omega{t})$を与えたときに、電荷密度が$\delta{P}=g\cos(2\omega{t})$のようには振動しないと仮定します
【ステップ2】光エネルギーを摂動と捉え、励起状態を複数の電子配置で表現する
次に、光エネルギーを摂動と捉えます。
摂動とは
量子力学・化学における摂動とは、全ての固有エネルギー$\epsilon_i$、固有関数$\phi_i$がわかっている状態$\hat{H}$があり、それに小さな外場、外乱$\hat{V}$が加わった場合の波動関数を$\psi=\sum_ic_i\phi_i$と表す手法です。
光エネルギーを摂動と捉えることで、基底状態の計算で得られた分子軌道を利用して励起電子状態を複数作成できるようになります。
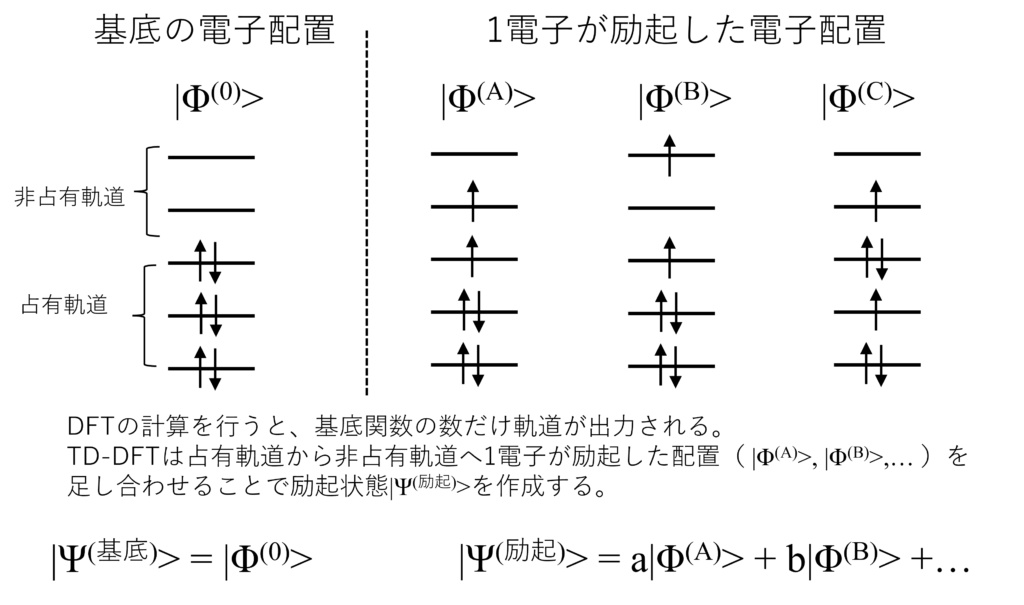
例えばホルムアルデヒド(電子$16$個)に対して6-31G(d,p)の基底関数を使うと、基底関数の数は$40$個です。
占有軌道は$8$個、非占有軌道は$32$個になるから、1電子配置は$8\times32=256$個作れるんだね。
TD-DFTはすでに得られている分子軌道を利用し、占有軌道から非占有軌道へ1電子が励起した配置($\ket{\Phi^{(A)}}$, $\ket{\Phi^{(B)}}$,… )を複数足し合わせることで励起状態$\ket{\Psi^{\text{励起}}}$を作成します。
つまり、$\ket{\Psi^{\text{励起}}}=a\ket{\Phi^{(A)}}+b\ket{\Phi^{(B)}}+\cdots$で励起状態を表します。
TD-DFTでは励起状態の計算を始める前に、基底状態の計算を行い、分子軌道を得ています。
【ステップ3】励起状態のエネルギーは発散しないとする
$\ket{\Psi^{\text{励起}}}=a\ket{\Phi^{(A)}}+b\ket{\Phi^{(B)}}+\cdots$の$a$、$b$の選び方なんて無限にあるんじゃないの?
励起エネルギーが発散しないと仮定すると$a$,$b$,…の選び方は有限になります。
TD-DFTでは、電磁波(摂動)を分子に当てた際に、励起エネルギーが発散しないと仮定して励起電子状態を求めます。これは「摂動に対する応答関数が励起エネルギーにおいて極をもつ」とも言われます。
この過程がないと、摂動に対する応答に$\exp(at)$の項が入り、時間発展とともに電子密度が発散するという化学的に意味のない解になってしまいます。
【まとめ】実際にどのような計算式を解けばよいか
では、ステップ1~ステップ3を利用するとどのような計算式で励起エネルギーを知ることができるかを紹介します。
結論的には次の行列(TD行列とします)の固有値を求めれば励起エネルギー$\omega$を知ることができます。(脱励起の効果を無視(Dancoff近似)して計算式を簡単にしています)
$\begin{pmatrix}{\epsilon}_{a}-\epsilon_{i}+\frac{\partial{F_{ai}}}{\partial{P_{ai}}}&\frac{\partial{F_{ai}}}{\partial{P_{bi}}}&\cdots\\\frac{\partial{F_{bi}}}{\partial{P_{ai}}}&{\epsilon}_{b}-\epsilon_{i}+\frac{\partial{F_{bi}}}{\partial{P_{ab}}}&\cdots\\\vdots&\vdots&\ddots\end{pmatrix}\boldsymbol{d}$$=\boldsymbol{\omega}\boldsymbol{d}$
ここで、$i,j,\cdots$は占有軌道、$a,b\cdots$は非占有軌道を表しているとします。$\epsilon_k$は$k$番目の軌道エネルギー、$\frac{\partial{F_{pq}}}{\partial{P_{rs}}}$は電子密度の変化時にフォック行列がどのように変化するかを表し、$\frac{\partial{F_{pq}}}{\partial{P_{rs}}}=(pq|rs)+\iint\phi_p^\ast\phi_q\frac{\delta^2E_{xc}}{\delta\rho(r)\delta\rho(r^\prime)}\phi^\ast_r\phi_s$で与えられます。
占有軌道が$N_\text{occ}$個、非占有軌道が$N_\text{var}$個あるとすると、$N_\text{occ}\times{N_\text{var}}$次元の連立方程式を解くことになります。
詳細なTD-DFT表式の導出はこちら


補足

【具体例】水の励起エネルギー
最後に、水分子を使ってTD行列を具体的に見ましょう。3重項の励起エネルギーを調べます。
TD-DFTでは多数のグリッド部分の数値積分が困難なので、TD-HFを利用し、基底関数は簡便なSTO-3Gとします。
STO-3Gの場合、占有軌道は$5$個、非占有軌道は$2$個になるので、$10\times10$の行列固有値を解くと励起エネルギーが得られます。
まずはRHF計算を行い、分子軌道の軌道係数$C_{\mu{p}}$を得ます。STO-3Gでは基底関数は$7$個になるので、得られる分子軌道も$7$個です。
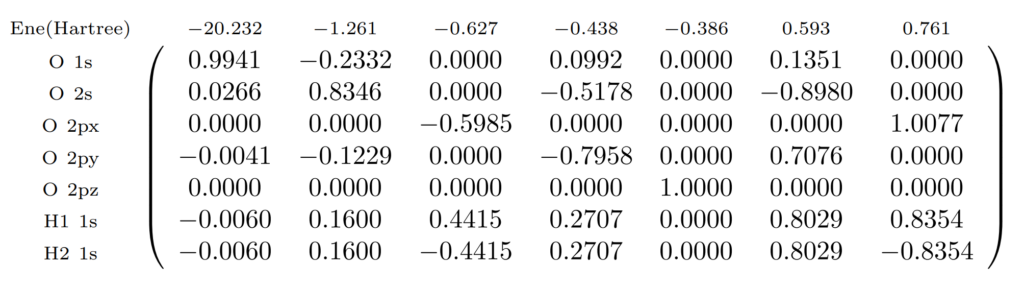
水分子の電子数は$10$個なので、占有軌道は$5$個、非占有軌道は$2$個になります。
TD-HFでもTD-DFTと同じように、TD行列の固有値問題を解けば励起エネルギーを得ることができます。$\begin{pmatrix}{\epsilon}_{a}-\epsilon_{i}+\frac{\partial{F_{ai}}}{\partial{P_{ai}}}&\frac{\partial{F_{ai}}}{\partial{P_{bi}}}&\cdots\\\frac{\partial{F_{bi}}}{\partial{P_{ai}}}&{\epsilon}_{b}-\epsilon_{i}+\frac{\partial{F_{bi}}}{\partial{P_{ab}}}&\cdots\\\vdots&\vdots&\ddots\end{pmatrix}\boldsymbol{d}$$=\boldsymbol{\omega}\boldsymbol{d}$
TD-HFでは$\frac{\partial{F_{pq}}}{\partial{P_{rs}}}$を解析的に与えられます。3重項励起の場合は$\frac{\partial{F_{pq}}}{\partial{P_{rs}}}=(pq|rs)-(pq|sr)$$=\sum_{\mu\nu\lambda\kappa}^\text{NBasis}C_{\mu{p}}C_{\nu{q}}C_{\lambda{r}}C_{\kappa{s}}\{(\mu\nu|\lambda\kappa)-(\mu\nu|\kappa\lambda)\}$です。($p,q,r,s$は分子軌道、$\mu\nu\lambda\kappa$は原子軌道で、$C_{\mu{p}}$は分子軌道$\phi_p$の軌道係数)
$5$個の占有軌道、$2$個の非占有軌道から得られる1電子励起の場合の数は$10$通りなので、TD行列は$10\times10$行列になります。

最後にTD行列の固有値を求めると$10$個の固有値が得られます。比較としてTD-DFTでも計算を行っています(ソフトウェアはGAMESS、汎関数はCAMB3LYP、基底関数はSTO-6G)(STO-3Gではエラーが起きました)
| 励起エネルギー(eV) | 1st | 2nd | 9th | 10th | |
| TD-HF | 11.8 | 14.7 | ・・・ | 546.0 | 547.9 |
| TD-DFT | 9.7 | 12.1 | 523.9 | 526.7 |
TD-HFとTD-DFTには数値の違いはあるものの、大まかな傾向は一致した結果を得ることができます。
このように、RHFで得られた分子軌道をもとにTD行列の固有値を求めることで励起エネルギーを知ることができるのです。
まとめ
このページでは、TD-DFTで分子の励起エネルギーが計算できる理由とそこに必要な仮定を解説し、最終的に詳細なTD-DFTの表式をご紹介しました。
化学者にとって分子の励起エネルギーは分子の色彩や吸収スペクトルを知るうえで非常に重要です。
TD-DFTを利用した励起エネルギーの計算はGAMESSやGaussianで簡単に計算できますので、皆さんも一度トライしてみてください。