

水分子などの分子動力学シミュレーション(MD)はヘリウムなどの単原子分子に比べてアルゴリズムの難易度が上がります。
しかし、MDでは水分子やタンパク質といった「形」があるもののシミュレーションのほうが単原子状分子のものよりも圧倒的に多いでしょう。
このページでは回転運動を行う粒子に対して分子動力学シミュレーションを行うためのアルゴリズムを紹介します。
- 分子に働く回転トルクを計算する方法を紹介します。
- 分子軸を回転させる方法を紹介します。
回転運動のある分子動力学シミュレーション
まずは水分子のMDを見てみましょう。
水分子のように並進運動に加えて回転運動もある分子の分子動力学シミュレーションは単原子状分子よりも華やかになります。
水350個の分子動力学シミュレーションです。
(VMDで可視化しています)

それぞれの水分子がいろいろな回転をしていてにぎやかだね。

でも、回転をプログラミングでどう表現するか想像できないや。。
回転運動のアルゴリズムには次の2ステップが必要です。

分子に働くトルクを計算して、分子の軸を回転させれば回転運動を表現できますよ。一緒に見ていきましょう。
分子回転アルゴリズムの詳細
分子に働くトルクを計算する
まずは、分子に働くトルクを計算するため、分子を構成する各原子$i$に働く力$\vec{F}$を計算しましょう。
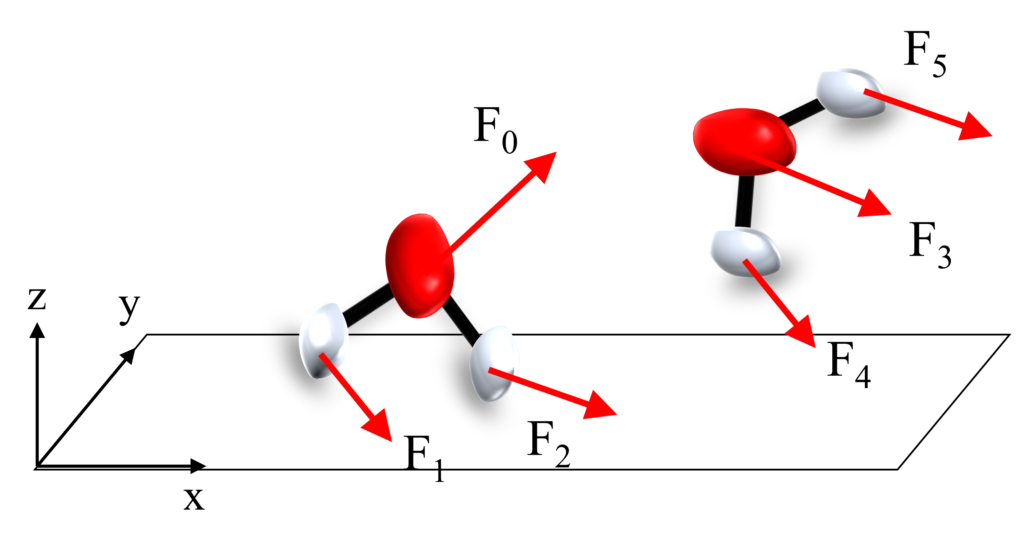
力$\vec{F}$は分子間力や静電ポテンシャルから計算できるね。
さて、いま得られている力$\vec{F}$は$F= \left(\begin{array}{ccc}F_x\\F_y\\F_z\end{array}\right)=\left(\begin{array}{ccc}2\\1\\3\end{array}\right)$のように書けますが、これは分子の回転軸と垂直になっていません。

トルクは力が回転軸と垂直になってないと計算できないよ?
そのため、得られている力$\vec{F}$の各分子軸方向の大きさを求めていきましょう。
力の分解で役立つのが正射影つまり内積の考え方です。例えば、単位ベクトル$\vec{d}$へ力$\vec{F}$を分解した$\vec{F_d}$は内積を使うと$\vec{F_d}=(\vec{F}\cdot\vec{d})\vec{d}$と書くことができます。
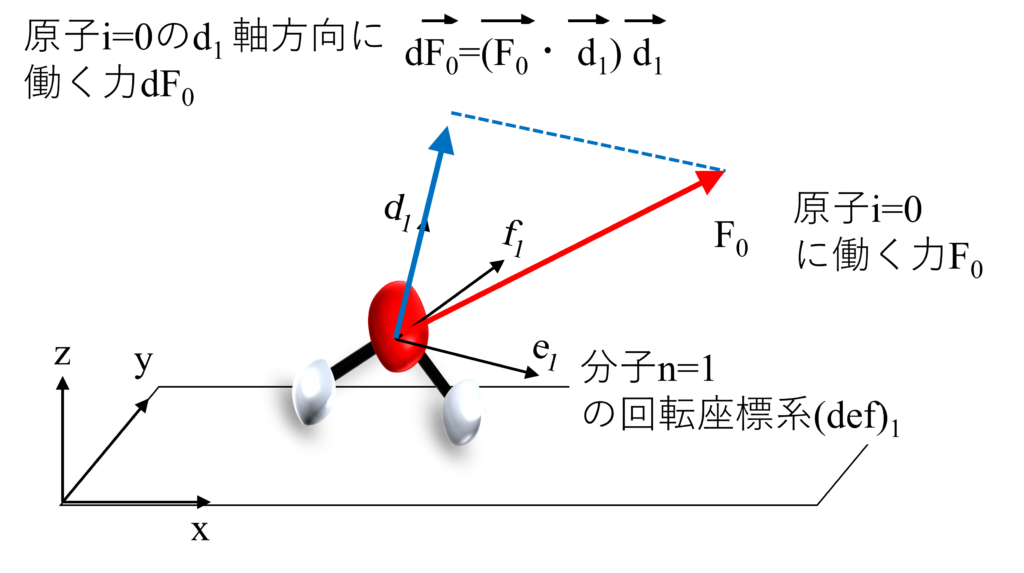
例として、ある分子の$d$軸が$\vec{d} = \left(\begin{array}{ccc}0.6\\0.8\\0\end{array}\right)$であったとき、$F=\left(\begin{array}{ccc}F_x\\F_y\\F_z\end{array}\right)=\left(\begin{array}{ccc}2\\3\\1\end{array}\right)$の$d$軸成分の大きさは$\vec{F}\cdot\vec{d} = \left(\begin{array}{ccc}2\\1\\3\end{array}\right)\cdot\left(\begin{array}{ccc}0.6\\0.8\\0\end{array}\right)=2$と、内積で簡単に計算できます。
内積を利用すると力を簡単に分解できるね。
実際のプログラミングでは、、、
回転運動をプログラミングする上で最も注意するべき点(つまづきやすい点)は「分子の回転軸$\vec{d}_n$もプログラミング上はxyz座標系の配列要素で計算を行う」だと思います。
例えば、$n$番目分子の$d$軸単位ベクトル$\vec{d_n}$もプログラミング上はxyz軸座標系のベクトル配列として表します。
例として、$n$番目の分子について、$I$番目の原子に働く力$F(NDim,I)$を$d$軸、$e$軸、$f$軸に分解した力$dF(I)$、$eF(I)$、$fF(I)$を考えてみましょう。
各軸方向の単位ベクトルを用いることでそれぞれ次のように計算できます。
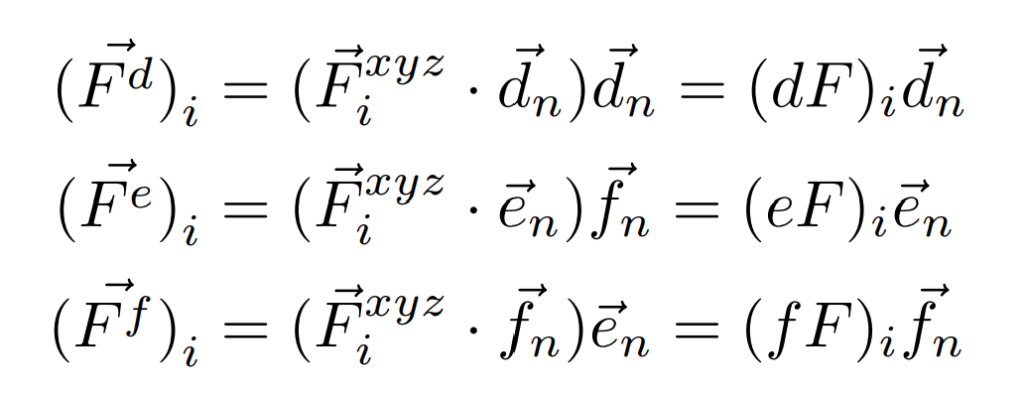
内積を利用することで、各軸方向の力の大きさ$dF(I)$、$eF(I)$、$fF(I)$は次のようにプログラミングできるのです。
dF(I)=F(1,I)*VEC_D(1,J)+F(2,I)*VEC_D(2,J)+F(3,I)*VEC_D(3,J)
eF(I)=F(1,I)*VEC_E(1,J)+F(2,I)*VEC_E(2,J)+F(3,I)*VEC_E(3,J)
fF(I)=F(1,I)*VEC_F(1,J)+F(2,I)*VEC_F(2,J)+F(3,I)*VEC_F(3,J)プログラミング上では常に$xyz$軸での値をメインに使っていったほうが見通しが楽なんだね。
$d$軸、$e$軸、$f$軸方向の力の大きさが分かったことで、各軸に働くトルク$T$は「腕の長さ」×「力の大きさ」で簡単に計算できます。
ベクトルで書くと$\vec{T}^{def}=\vec{r}^{def}\times\vec{F}^{def}$だね。
簡単な例として、酸素Oにe軸方向の力が働いている状況を考えてみましょう。
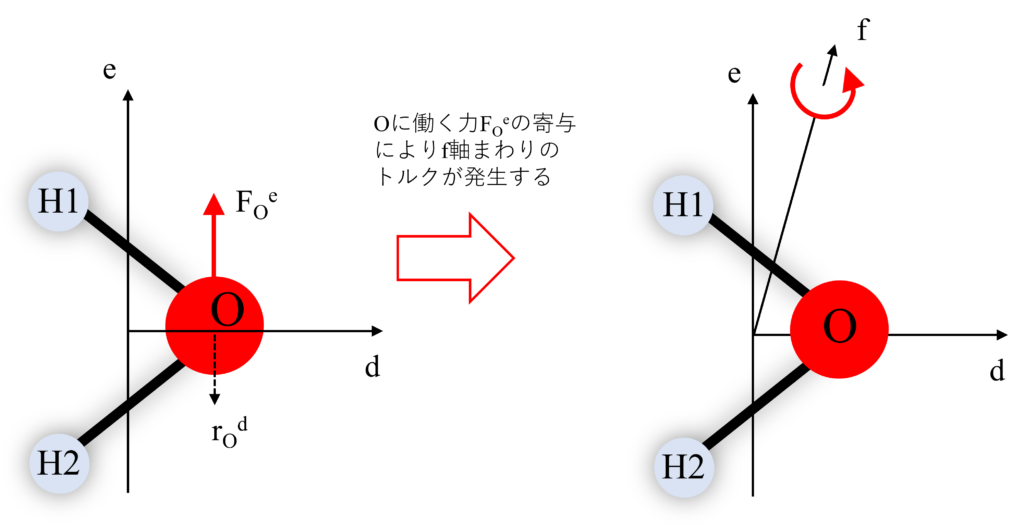
$e$軸方向へ酸素Oに力$F$が働くと「うでの長さ$r$」をかけたトルク$T=r{\times}F$が発生します。
結果として、水分子の$de$軸を反時計回り回そうとする原動力につながるんだね。
回転方程式を解き、分子の回転軸を回転させる
トルク$T$が計算できたことで、回転方程式$I\frac{\text{d}\omega}{\text{d}t}=T$から角運動量$\omega$の変化量を知ることができます。$I$は慣性モーメントです。
回転方程式は各軸ごとにあるよ。例えば$d$軸方向は$I^d\frac{\text{d}\omega^d}{\text{d}t}=T^d$だよ。
最後に微小時間に$def$軸がどれだけ回転するか分かれば各分子の位置を更新できます。
微小時間$\Delta{t}$の間には、$d$軸回りに$\omega^d\Delta{t}$、$e$軸回りに$\omega^e\Delta{t}$、$f$軸回りに$\omega^f\Delta{t}$回転するんだね。
$f$軸回りに$\omega^f\Delta{t}$だけ回転させたとき他の$de$軸がどのように変化するか見ていきましょう。
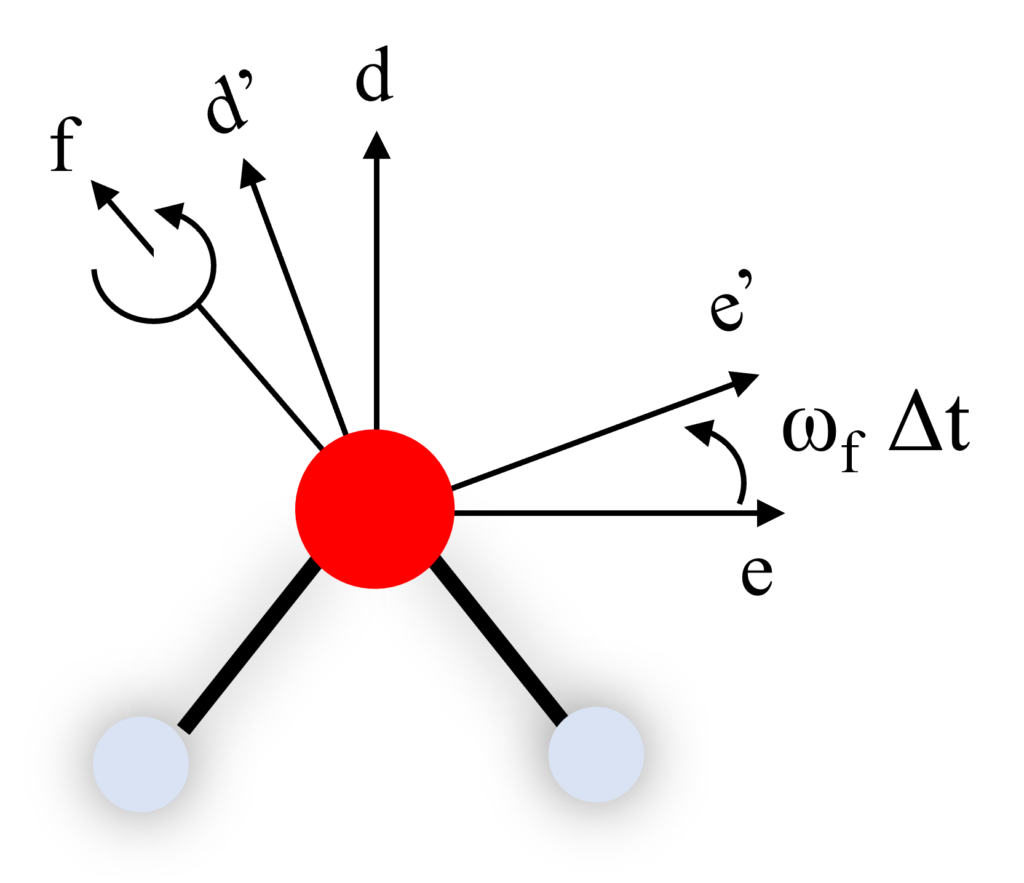
$f$軸を真上から見てみましょう。この$f$軸を中心に$de$軸を$\omega^f\Delta{t}$だけ回転させて$d^\prime{e^\prime}$軸に移るとき、$d^\prime{e^\prime}$軸は行列を用いて表すことができます。
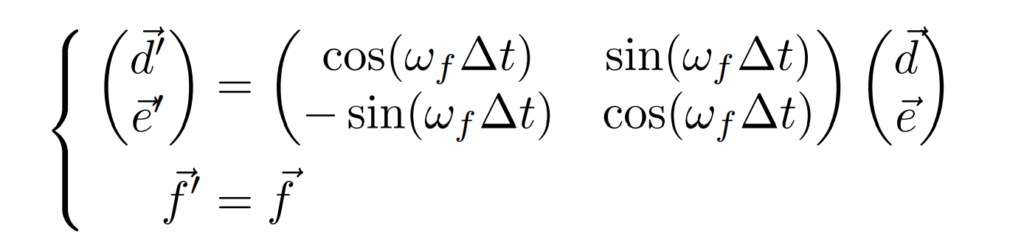
回転といえば複素数$i$をかけるって聞いたけど、行列でもできるの?
プログラミングの観点では、回転は行列で表現したほうが見通しを立てやすいかと思います。
実際のプログラミングでは、、、
プログラミングする上では「分子回転軸のベクトル$\vec{d}$もxyz座標系の配列要素で計算を行う」と見通しがたちやすいです。
例えば$N$番目分子における$d$軸の単位ベクトルもプログラミング上は、VEC_D(NDim,N)として扱います($\text{NDim}=1,2,3$がそれぞれ$x,y,z$軸)。
例えば、$N$番目分子の$de$座標を$f$軸回りに角度$dW(N)$回転させた場合、$\text{d}^\prime{\text{e}^\prime}$座標軸はつぎのようにプログラミングして得ることができます。
DO NDim = 1,3
VEC_D_P(NDim,N) = dcos(dW(N))*VEC_D(NDim,N) + dsin(dW(N))*VEC_E(NDim,N)
VEC_E_P(NDim,N) =-dsin(dW(N))*VEC_E(NDim,N) + dcos(dW(N))*VEC_E(NDim,N)
VEC_F_P(NDim,N) = VEC_F(NDim,N)
ENDDOこの操作を$f$軸、$e$軸、$d$軸と順番に行うと、最終的に$d$、$e$、$f$軸は$d^{\prime\prime\prime}$、$e^{\prime\prime\prime}$、$f^{\prime\prime\prime}$へ移ります。
同じ操作を$e$軸、$f$軸でもおこなうんだね。
$t+\Delta{t}$における分子座標系が$d^{\prime\prime\prime}$、$e^{\prime\prime\prime}$、$f^{\prime\prime\prime}$であることから、重心の運動と合わせると各原子の位置を更新することができるのです。
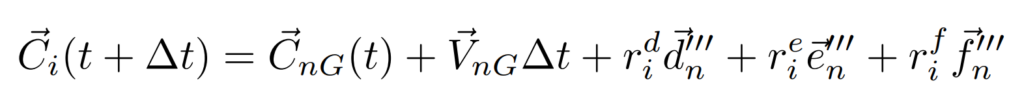
まとめると、1stステップでトルクを計算して、2ndステップで分子軸を回転させることで回転運動を表せたんだね。
まとめ
このページでは次のことを確認しました。
- 力を分解する際は内積を使うと簡単にできる。
- 回転行列を用いることで分子軸を簡単に回転させることができる。
- プログラミングする上ではdef系のベクトルでも常にxyz系での表記を意識する。
分子の回転を含む分子動力学シミュレーションを行うためにはその数値が実験座標系(xyz系)で記述されているのか、または分子回転座標系(def系)で記述されているのかを常に意識することが重要です。









